Pipeline Catalog: RNA Sequencing
Uploading Data
RNA Sequencing datasets should be uploaded as a collection of paired-end FASTQ files (gzip-compressed). There are two options for how the data can be formatted for upload:
- Use the sample IDs encoded in the FASTQ file names, or
- Use a samplesheet CSV to specify the sample IDs for each pair of FASTQs.
Parsing Sample IDs from FASTQ File Names
When parsing the sample IDs from the FASTQ file names, all of the following patterns can be used to assign the sample name ("SampleName" in the examples below) to a FASTQ file:
Note that each Read 1 file must have a matching Read 2 file, which differs only by 1 -> 2.
Pattern 1:
SampleName.R1.fastq.gz
┃ ┃┃ ┃ ┗━ Extension .gz is optional
┃ ┃┃ ┗━━━━━━ Extension can be '.fastq' <or> '.fq'
┃ ┃┗━━━━━━━━━ Read pair: 'R1' <or> '1' allowed (with matching 'R2' <or> '2')
┃ ┗━━━━━━━━━━ Separator can be '.' <or> '_'
┗━━━━━━━━━━━━━━━━━━━━ Sample identifier ('SampleName' in this case)
Pattern 2:
SampleName_S1_L001_R1_001.fastq.gz
┃ ┃ ┃ ┃┗━ Extension must be '_001.fastq.gz'
┃ ┃ ┃ ┗━━ Read pair: 'R1' (with matching 'R2')
┃ ┃ ┗━━━━━ Lane on Illumina sequencer
┃ ┗━━━━━━━━━━ Sample index number
┗━━━━━━━━━━━━━━━━━━━━━━ Sample identifier ('SampleName' in this case)
Pattern 3:
SampleName_S1_R1_001.fastq.gz
┃ ┃ ┃┗━━━━━━ Extension must be '_001.fastq.gz'
┃ ┃ ┗━━━━━━━ Read pair: 'R1' (with matching 'R2')
┃ ┗━━━━━━━━━━ Sample index number
┗━━━━━━━━━━━━━━━━━━━━━━ Sample identifier ('SampleName' in this case)
Organizing FASTQs with a Sample Sheet
The advantages of using a sample sheet when uploading data are:
- (a) the file names do not have to follow any of the patterns listed above,
- (b) additional sample metadata can be added en masse, and
- (c) reads from a single sample can be combined across multiple file pairs.
To use a sample sheet, simply create a file named samplesheet.csv in the
folder containing the data to be uploaded with the format:
sample,fastq_1,fastq_2
SampleA,SampleA.R1.fastq.gz,SampleA.R2.fastq.gz
SampleB,SampleB.R1.fastq.gz,SampleB.R2.fastq.gz
Note:
- File names do not need to match any particular pattern
Multiple FASTQ pairs can be listed for any sample
Any additional metadata can be added as columns to the sample sheet. For example, gene expression analysis with the nf-core/rnaseq pipeline can use information on
strandednessfor each sample, as described below.
RNAseq (nf-core/rnaseq)
![]()
nf-core/rnaseq is a best-practice pipeline for RNA sequencing analysis using STAR, RSEM, HISAT2 or Salmon with gene/isoform counts and extensive quality control.
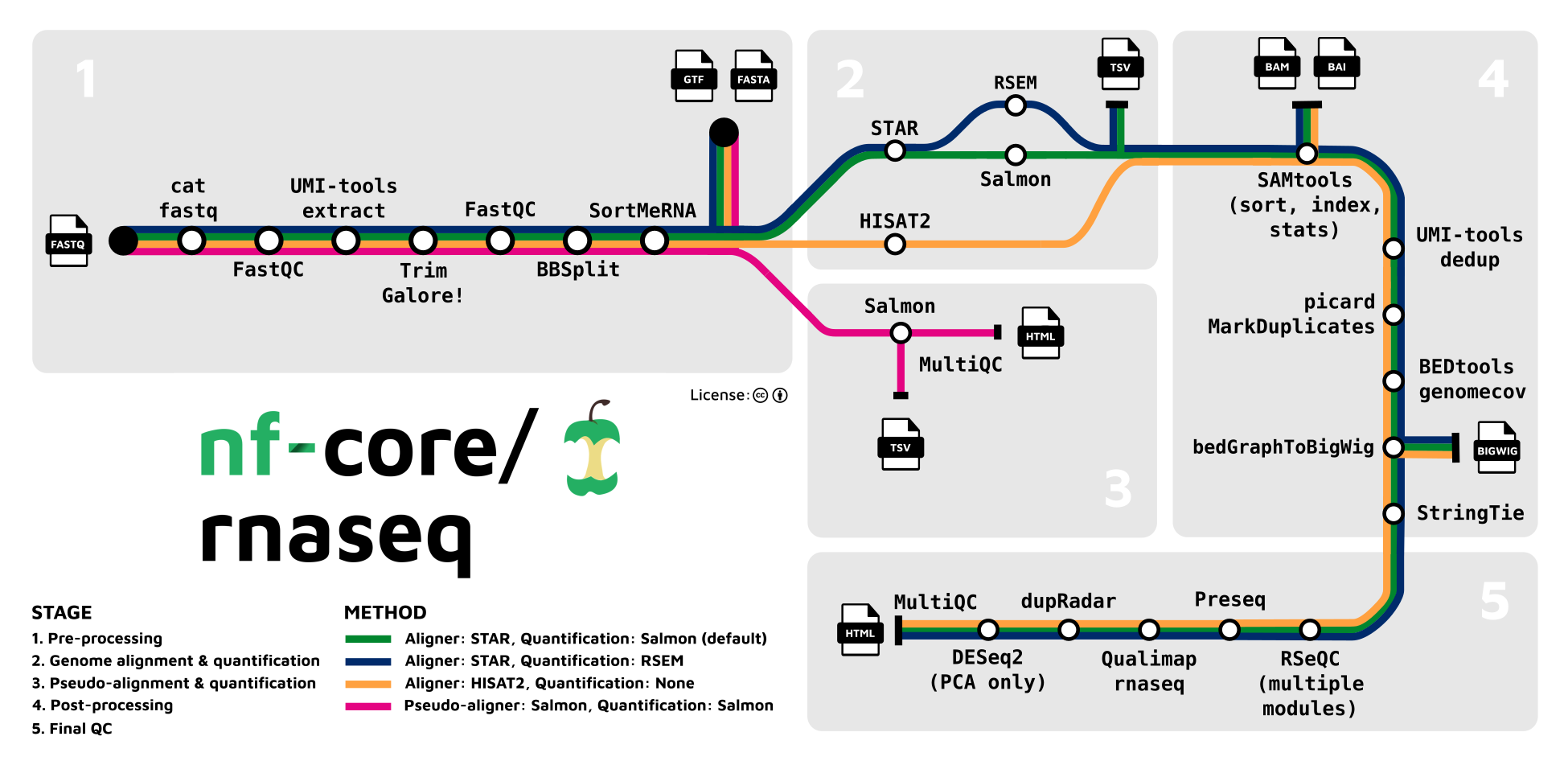
After adapter removal and quality trimming, reads are aligned to a reference genome with the STAR aligner and transcript abundances are estimated with either Salmon or RSEM (as selected by the user). Quality control metrics are computed for both the input reads and the transcript alignments, generating a single quality summary document via MultiQC.
Supports combining input datasets in a single analysis.
Supporting Media
User Guide
Quantification of gene expression requires a reference genome with a model of what genes are encoded and their physical coordinates on the genome. A collection of curated reference genomes are provided using the AWS iGenomes, which is a standard approach for nf-core workflows.
To use a custom genome:
- Upload the genome's nucleotide sequence as a
Reference Genome (FASTA)reference - Upload the genome's gene coordinates as a
Reference Genome Features (GTF)reference - Launch the Gene Expression workflow, selecting those references in the
Reference Genome (Custom)section - If your GTF files is in GENCODE format, make sure to select that option
Custom Genome GTF Format:
The genome annotation format expected by the Salmon aligner is not particularly flexible, and so it can be challenging sometimes to provide a properly formatted GTF file.
As a point of reference, the NCBI GRCh38 reference file is formatted as follows:
chr1 BestRefSeq exon 11874 12227 . + . gene_id "DDX11L1"; gene_name "DDX11L1"; transcript_id "rna0"; tss_id "TSS31672";
chr1 BestRefSeq exon 12613 12721 . + . gene_id "DDX11L1"; gene_name "DDX11L1"; transcript_id "rna0"; tss_id "TSS31672";
chr1 BestRefSeq exon 13221 14409 . + . gene_id "DDX11L1"; gene_name "DDX11L1"; transcript_id "rna0"; tss_id "TSS31672";
chr1 BestRefSeq exon 14362 14829 . - . gene_id "WASH7P"; gene_name "WASH7P"; transcript_id "rna1"; tss_id "TSS15911";
chr1 BestRefSeq exon 14970 15038 . - . gene_id "WASH7P"; gene_name "WASH7P"; transcript_id "rna1"; tss_id "TSS15911";
chr1 BestRefSeq exon 15796 15947 . - . gene_id "WASH7P"; gene_name "WASH7P"; transcript_id "rna1"; tss_id "TSS15911";
chr1 BestRefSeq exon 16607 16765 . - . gene_id "WASH7P"; gene_name "WASH7P"; transcript_id "rna1"; tss_id "TSS15911";
Notably:
- Genes are described in terms of
exonrecords - Each record contains fields for
gene_id,gene_name,transcript_id, andtss_id
Reusing Custom Genome References
The first step in analyzing a custom genome reference is to compute the alignment index.
To avoid repeating this time-consuming step in subsequent analyses,
the user can select the previously-computed reference under the Reference Genome (Custom)
option when running the Gene Expression workflow.
Strandedness
When samples are prepared for RNA sequencing, the resulting molecular libraries can be designed to provide
information from either the positive strand, the negative strand, or both.
By default, the RNAseq analysis pipeline will automatically infer the strandedness of the input data.
However to explicitly define the strandedness of each sample the user can provided a strandedness
annotation when uploading data.
In that case, the values used for the strandedness column may be forward, reverse, or unstranded.
Parameters:
- Genome: Select the most appropriate reference genome for the experimental organism. All genomes are sourced from the iGenomes collection
- Read Trimming: By default, sequencing adapters are removed from the reads prior to sequence alignment. Depending on the way in which the sequencing library was prepared, the user may select the option to skip adapter trimming and/or remove additional bases from the 5' or 3' end of each read
- Alignment Options: By default, reads are aligned first with the STAR aligner and then transcript abundances are estimated with Salmon. The user may alternately choose to use the RSEM or HISAT2 aligners
- Process Skipping: In addition to performing gene and transcript quantification, a number of optional processes may be skipped by the user. The Qualimap and dupRadar processes are skipped by default because of the relatively large amount of resources they utilize
- UMI Options: If UMIs were used for controlling PCR duplicates, the user may specify the string or regex pattern which may be used to extract the UMI from the sequence read
- Read Filtering: The BBSplit and SortMeRNA methods for removal of contaminating sequences are disabled by default and may be enabled by the user
Workflow Repository: github.com/nf-core/rnaseq
Citations:
- nf-core/rnaseq: Harshil Patel, Phil Ewels, Alexander Peltzer, Rickard Hammarén, Olga Botvinnik, Gregor Sturm, Denis Moreno, Pranathi Vemuri, silviamorins, Lorena Pantano, Mahesh Binzer-Panchal, BABS-STP1, nf-core bot, FriederikeHanssen, Maxime U. Garcia, James A. Fellows Yates, Chris Cheshire, rfenouil, Jose Espinosa-Carrasco, … George Hall. (2022). nf-core/rnaseq: nf-core/rnaseq v3.8.1 - Plastered Magnesium Mongoose (3.8.1). Zenodo. https://doi.org/10.5281/zenodo.6587789
- nf-core: Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020 Mar;38(3):276-278. doi: 10.1038/s41587-020-0439-x. PubMed PMID: 32055031.
- STAR: Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner Bioinformatics. 2013 Jan 1;29(1):15-21. doi: 10.1093/bioinformatics/bts635. Epub 2012 Oct 25. PubMed PMID: 23104886; PubMed Central PMCID: PMC3530905.
- Salmon: Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression Nat Methods. 2017 Apr;14(4):417-419. doi: 10.1038/nmeth.4197. Epub 2017 Mar 6. PubMed PMID: 28263959; PubMed Central PMCID: PMC5600148.
- RSEM: Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome BMC Bioinformatics. 2011 Aug 4;12:323. doi: 10.1186/1471-2105-12-323. PubMed PMID: 21816040; PubMed Central PMCID: PMC3163565.
Immune Clonotypes
Independently from gene expression analysis, bulk RNA sequencing can be used to reconstruct the lineages of B- and T- cells present in a sample by de novo reconstruction of BCR and TCR loci. The TRUST4 tool used in this workflow is described by the authors as:
A computational tool to analyze TCR and BCR sequences using unselected RNA sequencing data, profiled from solid tissues, including tumors. TRUST4 performs de novo assembly on V, J, C genes including the hypervariable complementarity-determining region 3 (CDR3) and reports consensus of BCR/TCR sequences. TRUST4 then realigns the contigs to IMGT reference gene sequences to report the corresponding information. TRUST4 supports both single-end and paired-end sequencing data with any read length.
User Guide
Workflow Repository: github.com/FredHutch/pw-bulk-rna-immune-clonotypes
Citations:
- TRUST4: Song L, Cohen D, Ouyang Z, Cao Y, Hu X, Liu XS. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat Methods. 2021 Jun;18(6):627-630. doi: 10.1038/s41592-021-01142-2. Epub 2021 May 13. PMID: 33986545.
RNA Fusion (nf-core/rnafusion)
![]()
nf-core/rnafusion is a workflow for detecting gene fusions in the transcriptome of cancer patients, identifying clinically actionable targets for precision oncology. The workflow accepts as inputs RNA-sequencing data and currently supports alignment against the latest human reference genome.
Please note that whilst sharing some similarities, this workflow is independent of nf-core/rnaseq.
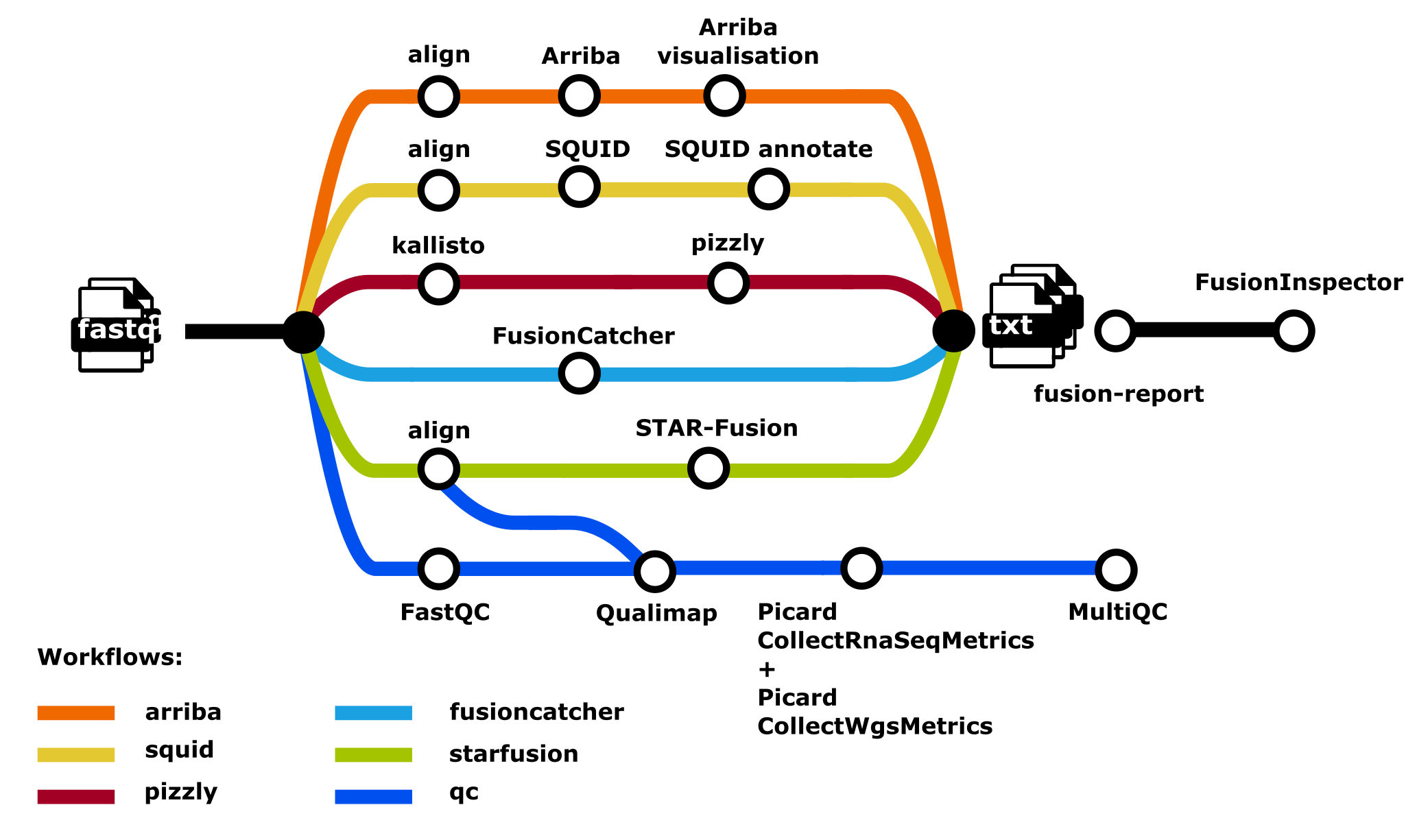
Supporting Media
User Guide
Parameters
- Reference Genome:
nf-core/rnafusiononly supportsGRCh38 - Fusion Detection Tools:
| Tool | Single-end reads |
|---|---|
| Arriba | No |
| FusionCatcher | Yes |
| Pizzly | No |
| Squid | No |
| STAR-Fusion | Yes |
- Report Generation: Collate results from fusion detection tools into a HTML report (highly recommended). This parameter can be selected in conjunction with any of the options selected in the
Fusion Detection Toolsparameter. - Read Trimming: Perform automated read trimming using
BBMAP. It is recommended to run the standaloneFastQCworkflow prior tonf-core/rnafusionto determine if your sequencing data requires the use of this parameter.
Workflow Repository: github.com/nf-core/rnafusion
Citations:
-
Martin Proks, Maxime Garcia, Rickard Hammarén, Phil Ewels, Chadi Saad, Matthias Stahl, & Szilveszter Juhos. (2020). nf-core/rnafusion: nf-core/rnafusion:1.2.0 (1.2.0). Zenodo: doi.org/10.5281/zenodo.3946477
-
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen. Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
HLA Typing (arcasHLA)
Human leukocyte antigen (HLA) typing is a genetic test that identifies protein markers on your cells that help your immune system tell the difference between your cells and foreign cells.
The six main HLA markers are: HLA-A, HLA-B, HLA-C, HLA-DR, HLA-DP, HLA-DQ.
HLA typing is used to match patients and donors for bone marrow, cord blood, or organ transplants. It also screens transplant recipients for antibodies that might target the donated tissue or organ.
The arcasHLA tool infers HLA genotypes from bulk RNA sequencing data.
Supports combining input datasets in a single analysis.
User Guide
Reference Database: The arcasHLA tool uses the IPD-IMGT/HLA Database as a reference for HLA sequences. The user may select a specific database version if needed.
Analysis Parameters: The following parameters may be selected by the user:
genes: The specific set of HLA genes to be reported (default: all)population: Sample population (default: prior)zygosity_threshold: Threshold for ratio of minor to major allele nonshared count to determine zygosity (default: 0.15)min_count: Minimum gene read count required for genotyping (default: 75)
Tool Repository: github.com/RabadanLab/arcasHLA
Workflow Repository: github.com/CirroBio/nf-arcashla
Citations:
Orenbuch, Rose et al. “arcasHLA: high-resolution HLA typing from RNAseq.” Bioinformatics (Oxford, England) vol. 36,1 (2020): 33-40. doi:10.1093/bioinformatics/btz474
Nanostring nCounter (nf-core/nanostring)
[]
The nf-core/nanostring pipeline allows the analysis of NanoString data. The pipeline performs quality control, normalization, and annotation of the obtained counts.
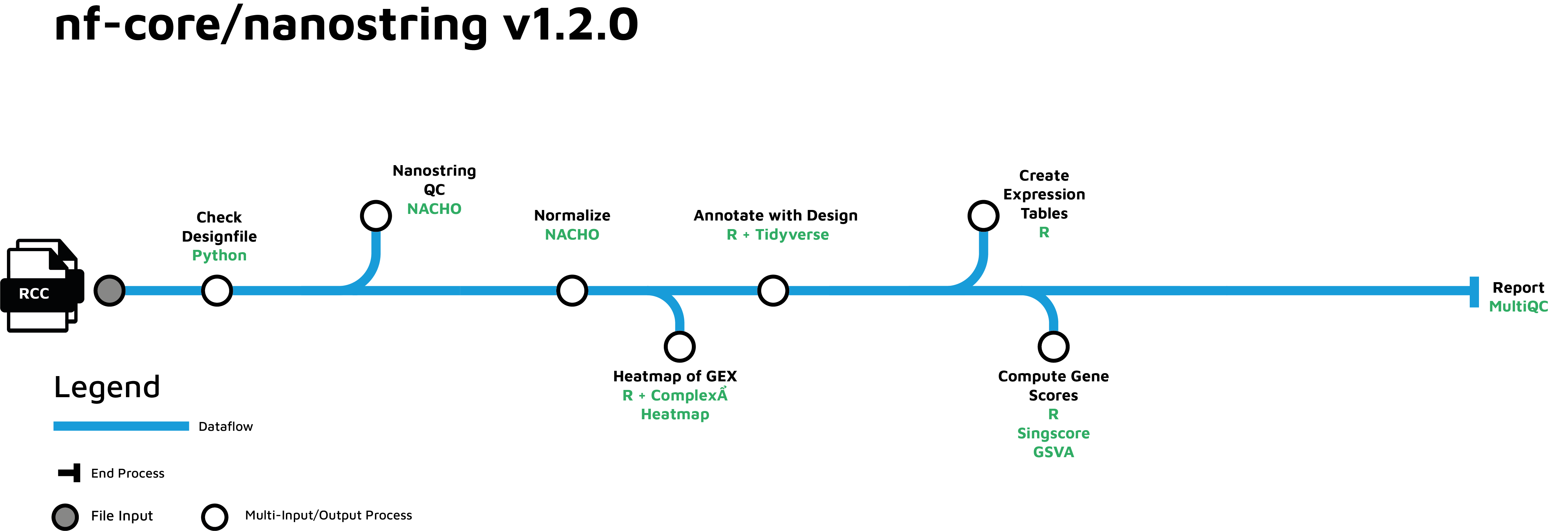
Metadata
To use a sample label in the heatmap display produced by the pipeline which is distinct
from the sample IDs embedded in the filenames, upload a samplesheet.csv file alongside
the input RCC files.
For example:
sample,file,OTHER_LABEL
sampleA,sampleA.RCC,Timepoint 1
sampleB,sampleB.RCC,Timepoint 2
sampleC,sampleC.RCC,Timepoint 3
sampleD,sampleD.RCC,Timepoint 4
When running the analysis, the "Heatmap ID Column" parameter can be used to indicate the column in the samplesheet that should be used for the heatmap display.
Note that the
samplecolumn provided in thesamplesheet.csvfile will be referenced by the nf-core/nanostring pipeline asSAMPLE_ID.
Tool Repository: github.com/nf-core/nanostring