Pipeline Catalog: DNA Sequencing
Uploading FASTQ Data
DNA Sequencing datasets should be uploaded as a collection of paired-end FASTQ files (gzip-compressed). There are two options for how the data can be formatted for upload:
- Use the sample IDs encoded in the FASTQ file names, or
- Use a samplesheet CSV to specify the sample IDs for each pair of FASTQs.
Parsing Sample IDs from FASTQ File Names
When parsing the sample IDs from the FASTQ file names, all of the following patterns can be used to assign the sample name ("SampleName" in the examples below) to a FASTQ file:
Note that each Read 1 file must have a matching Read 2 file, which differs only by 1 -> 2.
Pattern 1:
SampleName.R1.fastq.gz
┃ ┃┃ ┃ ┗━ Extension .gz is optional
┃ ┃┃ ┗━━━━━━ Extension can be '.fastq' <or> '.fq'
┃ ┃┗━━━━━━━━━ Read pair: 'R1' <or> '1' allowed (with matching 'R2' <or> '2')
┃ ┗━━━━━━━━━━ Separator can be '.' <or> '_'
┗━━━━━━━━━━━━━━━━━━━━ Sample identifier ('SampleName' in this case)
Pattern 2:
SampleName_S1_L001_R1_001.fastq.gz
┃ ┃ ┃ ┃┗━ Extension must be '_001.fastq.gz'
┃ ┃ ┃ ┗━━ Read pair: 'R1' (with matching 'R2')
┃ ┃ ┗━━━━━ Lane on Illumina sequencer
┃ ┗━━━━━━━━━━ Sample index number
┗━━━━━━━━━━━━━━━━━━━━━━ Sample identifier ('SampleName' in this case)
Pattern 3:
SampleName_S1_R1_001.fastq.gz
┃ ┃ ┃┗━━━━━━ Extension must be '_001.fastq.gz'
┃ ┃ ┗━━━━━━━ Read pair: 'R1' (with matching 'R2')
┃ ┗━━━━━━━━━━ Sample index number
┗━━━━━━━━━━━━━━━━━━━━━━ Sample identifier ('SampleName' in this case)
Organizing FASTQs with a Sample Sheet
The advantages of using a sample sheet when uploading data are:
- (a) the file names do not have to follow any of the patterns listed above,
- (b) additional sample metadata can be added en masse, and
- (c) reads from a single sample can be combined across multiple file pairs.
To use a sample sheet, simply create a file named samplesheet.csv in the
folder containing the data to be uploaded with the format:
sample,fastq_1,fastq_2
SampleA,SampleA.R1.fastq.gz,SampleA.R2.fastq.gz
SampleB,SampleB.R1.fastq.gz,SampleB.R2.fastq.gz
Note:
- File names do not need to match any particular pattern
Multiple FASTQ pairs can be listed for any sample
Any additional metadata can be added as columns to the sample sheet. For example, paired somatic variant calling with the nf-core/sarek pipeline expects columns
patient,sex, andstatus, as described below.
Uploading BAM Data
Raw sequence data may also be uploaded in unaligned BAM format (uBAM). One of the benefits of using this format (as mentioned in the GATK discussion forum) is that provides a mechanism for attaching additional metadata to the sequence reads as needed.
The unaligned BAM file is most commonly the starting-point for analysis tools from the Genome Analysis Toolkit (GATK).
When uploading BAM files to Cirro, the sample name will be mapped directly
from the file name (removing the .unmapped.bam or .bam suffix).
To provide a custom set of sample names for a set of BAM files, use
the samplesheet method described above
(using the column name file instead of fastq_1).
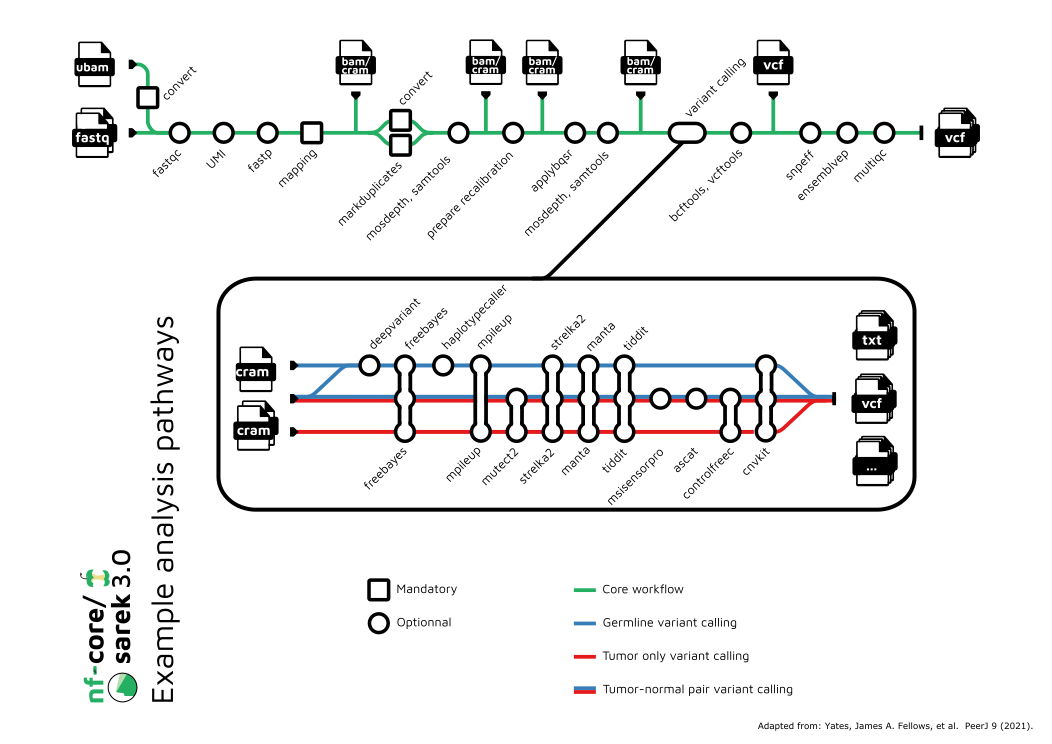
DNA Variant Calling (nf-core/sarek)
![]()
One of the most successful and productive open-science collaborations in the bioinformatics field is nf-core, a community effort to collect a curated set of analysis pipelines built using Nextflow. In addition to maintaining a robust catalog of community-generated analysis pipelines, the nf-core community effort has also been published in Nature Biotechnology.
nf-core/sarek is a bioinformatics workflow for detecting small (single nucleotide variants, insertions and deletions) and large scale (copy number alterations, structural variants, microsatellite instability) genomic variants in whole genome sequencing (WGS), whole exome sequencing (WES) or targeted gene panels.
Supports combining input datasets in a single analysis.
Supporting Media
User Guide
By default, the workflow follows GATK best practices preprocessing steps - samples are trimmed to discard low quality reads and aligned to the reference genome using BWA-MEM, PCR duplicates are marked and machine learning algorithms are applied to detect systematic errors made by the sequencer during base calling, allowing for confident calling of variant alleles at a genomic loci. Subsequent to preprocessing, the workflow diverges into one of three analysis modes discussed below. All variants are annotated for potential functional effects using VEP and snpEff, producing a detailed variant call format (VCF) file ready for interpretation by researchers/clinicians.
Analysis Modes
Sarek offers three analysis modes when running the workflow:
Germline variant calling: this analysis involves comparing a patient's normal sample (typically derived from blood or saliva) against the reference genome to identify causal variants in rare mendelian diseases.Paired somatic variant calling: the preferred method of somatic variant calling, a patient's tumor sample is assessed for the presence of variants that are absent in both the matched normal sample and reference genome to identify mutations driving tumor proliferation.Tumor only somatic variant calling: involves calling somatic variants in a patient's tumor sample in the event a matched normal is absent. Please note that due to the absence of a matched normal sample, results will contain germline variants. At best, this is an exploratory tool allowing researchers to detect the presence or absence of a mutation in a tumor sample.
Alignment vs. Variant Calling
The process of calling genomic variants from high-throughput sequencing data consists of two over-arching steps: sequence alignment and variant calling.
- In the sequence alignment stage a set of FASTQ files (or unmapped BAM files) are transformed into a set of mapped BAM files, in which each sequence fragment has been assigned a position in the reference genome of interest.
- In the variant calling stage the mapped BAM file is processed to identify any regions in the genome which appear to be significantly different from the reference.
In some cases the variant calling stage may be performed multiple times
with varying parameters in order to better interrogate the potential
variants in a particular sample.
To support this approach to data analysis, the nf-core/sarek variant calling
can be run in two different ways:
- End-to-End: FASTQ inputs can taken all the way through the alignment
and variant calling step, using the
Variant Calling (nf-core/sarek)pipeline, or - Modular: FASTQ inputs can be aligned against a reference genome with the
Align Reads (nf-core/sarek)pipeline, and then the resulting BAM files can be taken through the subsequentVariant Calling (nf-core/sarek)pipeline.
In this manner, a single dataset can be taken through multiple rounds of variant calling without having to repeat the time-consuming process of alignment.
Required Files
The only files which are required for running Sarek are the paired FASTQ files produced from the Illumina sequencer.
Those files may be uploaded to Cirro using either the graphical user interface or the command-line interface.
When prompted to indicate the analysis type, select Paired DNA-Seq.
Note: Before uploading files make sure to review the section on Sample Metadata below.
Sample Metadata
When performing Paired somatic variant calling, you must provide Sarek with the information necessary to compare each
patient's tumor sample against the matched normal control sample.
For this purpose, Sarek requires the following fields of metadata:
patient: A custom patient ID designating the patient/subject. A patient can have multiple samples, e.g sequenced across multiple lanes, and/or a tumor-normal pair.sex: One ofXX,XY, orNA. This is only taken into account when performing copy number alteration analysis in a tumor-normal pair.status: One of0or1denoting that a sample is normal or tumor, respectively.
Sample Metadata Option 1: Manual Entry
If you feel comfortable with JSON files, you can follow the instructions below to edit the JSON schemas for metadata. If not, contact the Cirro team for assistance.
To manually enter the sample metadata directly in the web-based user interface, simply
add the following fields (patient, sex, and status) to the metadata.schema.json
for your project as shown below:
{
"$schema": "http://json-schema.org/draft-07/schema#",
"type": "object",
"properties": {
"patient": {
"type": "string"
},
"sex": {
"type": "string",
"enum": [
"NA",
"XX",
"XY"
]
},
"status": {
"type": "integer",
"enum": [
0,
1
]
}
}
}
After updating the metadata schema, you will have the option to fill in the appropriate values for the samples which have been uploaded as part of your dataset.
Sample Metadata Option 2: Bulk Upload
To quickly annotate a large group of files you can upload a samplesheet.csv file in conjunction with your FASTQ files containing all of the metadata associated with your experiment.
This option can also be helpful if your file names do not follow the naming convention described above.
The presence of a samplesheet.csv file overrides Cirros automatic sample ID generation. Because of this, you must include sample ID information in the file in addition to the FASTQ file pairs.
In addition to the patient, sex, and status fields described above, the samplesheet.csv must include:
sample: A custom sample ID for each tumor and/or normal sample, more than one sample for each patient/subject is possible. Not to be confused withpatient. For example, if a patients normal sample was sequenced across 3 lanes (producing 6 FASTQ pairs), a common ID forsamplewill result in these files being merged post alignment.fastq_1: Read 1 FASTQ file name. Must contain the string*_R1.fastq_2: Read 2 FASTQ file name. Must contain the string*_R2.
For example:
patient,sex,status,sample,fastq_1,fastq_2
patient1,XX,0,patient1_normal,test_L001_R1.fastq.gz,test_L001_R2.fastq.gz
patient1,XX,0,patient1_normal,test_L002_R1.fastq.gz,test_L002_R2.fastq.gz
patient1,XX,0,patient1_normal,test_L003_R1.fastq.gz,test_L003_R2.fastq.gz
patient1,XX,1,patient1_tumor,test2_L001_R1.fastq.gz,test2_L001_R2.fastq.gz
patient1,XX,1,patient1_tumor,test2_L002_R1.fastq.gz,test2_L002_R2.fastq.gz
patient1,XX,1,patient1_tumor,test2_L003_R1.fastq.gz,test2_L003_R2.fastq.gz
Note: After uploading a
samplesheet.csvyou can still edit the metadata in Cirro using themetadata.schema.jsonfile provided above.
Capture Kits
In the event your sequencing data is targeted (whole exome or gene panel), you are required to upload a capture kit file containing the genomic coordinates of the exons included in the assay. This file should be in BED format, an example is given below:
chr1 69090 70008
chr1 621095 622034
chr1 861321 861393
chr1 865534 865716
chr1 866418 866469
Uploading Capture Kits
In Cirro, select References on the side menu and select + Add Reference. Select Genome Regions (BED) from the Reference Type dropdown menu. Name the file, and drag and drop your bed file into the portal. When you run your analysis, the file will be present under the Genomic Intervals dropdown menu.
If you do not have a capture kit, please contact a member of the Data Core and we will determine the best publicly available capture kit for your analysis.
Workflow Parameters
Reference Genome: Select the appropriate reference genome for your analysis.Whole Exome/Targeted Gene Panel Assay: Check this box if your sequencing data is targeted. This parameter will subset your variants to the regions specified in theGenomic Intervalsfile.Genomic Intervals: Select an intervals file from the dropdown menu. This parameter is required whenWhole Exome/Targeted Gene Panel Assayis selected. It is highly recommended to select a 'wgs_calling_regions' file for GRCh37/GRCh38 when performing a WGS analysis to perform a the analysis in parallelized chunks, greatly reducing the runtime and compute requirements.Variant Calling Type: Select one of 'Germline Variant Calling' or 'Somatic Variant Calling'. This parameter will toggle the variant calling tools available for your analysis.Variant calling tool(s): Select variant calling tools to include in your analysis. See the table below for the list of tools available for eachVariant Calling Type, and assay type.Variant Annotation: Select VEP and/or snpEff to produce functionally annotated variants.Plugins: 4 additional annotation plugins are available to compliment VEP.
| Tool | WGS | WES | Panel | Normal | Tumor | Somatic |
|---|---|---|---|---|---|---|
| DeepVariant | X | X | X | X | - | - |
| FreeBayes | X | X | X | X | X | X |
| GATK HaplotypeCaller | X | X | X | X | - | - |
| GATK Mutect2 | X | X | X | - | X | X |
| mpileup | X | X | X | X | X | - |
| Strelka2 | X | X | X | X | X | X |
| Manta | X | X | X | X | X | X |
| TIDDIT | X | X | X | X | X | X |
| ASCAT | X | X | - | - | - | X |
| CNVKit | X | X | - | X | X | X |
| Control-FREEC | X | X | X | - | X | X |
| MSIsensorPro | X | X | X | - | - | X |
Workflow Repository: github.com/nf-core/sarek
Citations:
-
Garcia M, Juhos S, Larsson M et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants [version 2; peer review: 2 approved] F1000Research 2020, 9:63 doi: 10.12688/f1000research.16665.2.
-
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen. Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
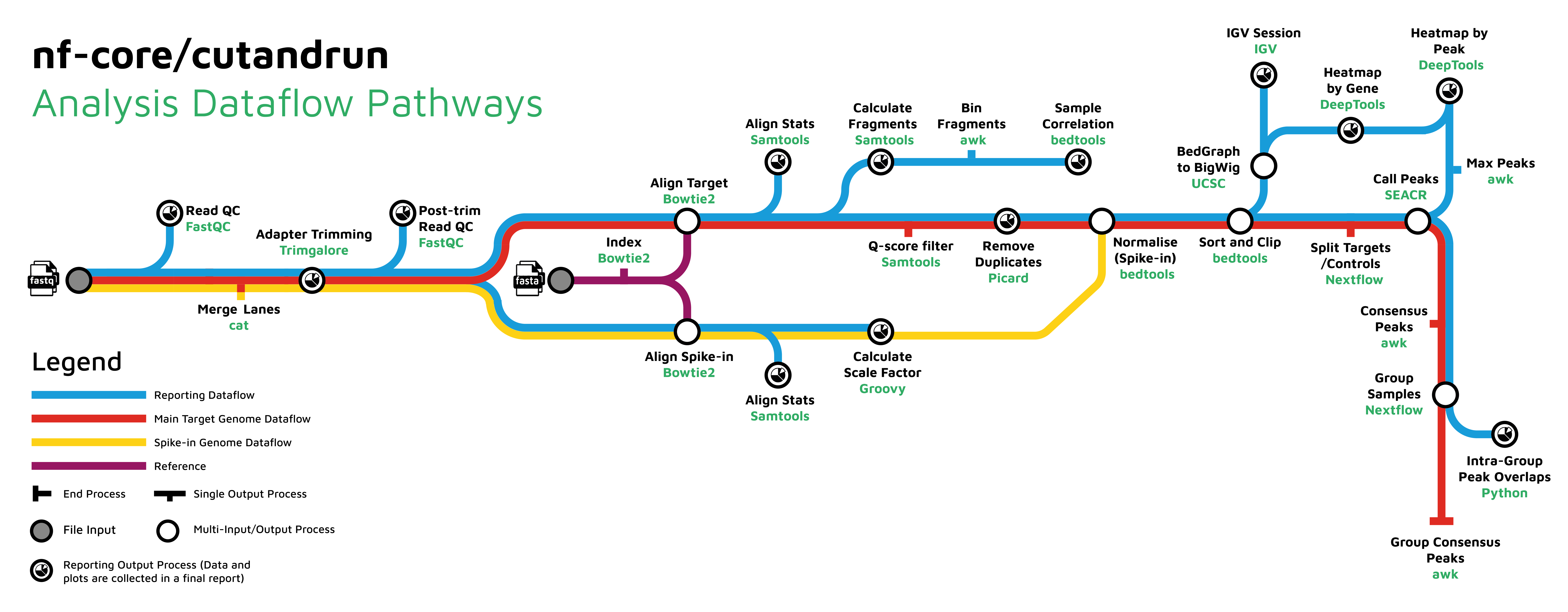
CUT&RUN (nf-core/cutandrun)
![]()
nf-core/cutandrun is a best-practice bioinformatic analysis pipeline for CUT&RUN and CUT&Tag experimental protocols that were developed to study protein-DNA interactions and epigenomic profiling.
The original CUT&Tag analysis protocol was developed by the Henikoff Lab at the Fred Hutch Cancer Center. The nf-core/cutandrun workflow was adapted from that protocol, originally written by Chris Cheshire (@chris-cheshire) and Charlotte West (@charlotte-west) from Luscombe Lab at The Francis Crick Institute, London, UK.
Supporting Media
User Guide
Sample Metadata: Control Annotation (e.g. IgG):
If you feel comfortable with JSON files, you can follow the instructions below to edit the JSON schemas for metadata. If not, contact the Cirro team for assistance.
After uploading paired-end DNA sequencing data in FASTQ format, samples must be annotated by experimental group and replicate. In order to analyze experiments in which an antibody control (e.g. IgG) has been used for normalization, each non-control group must be matched with the appropriate control group which it should be compared against.
To indicate the appropriate sample groups, first add fields for grouping, control, and replicate
to the metadata.schema.json for the project as shown here:
{
"$schema": "http://json-schema.org/draft-07/schema#",
"type": "object",
"properties": {
"grouping": {
"type": "string",
"description": "Used to indicate groups of samples which are replicates of the same experimental condition"
},
"replicate": {
"type": "integer",
"description": "Used to indicate the experimental replicate within each experimental condition"
},
"control": {
"type": "string",
"description": "Used to indicate which 'grouping' should be used as a matched control"
}
}
}
Next, update those metadata fields (grouping, replicate, and control) for the uploaded data.
If no antibody controls were used in the experiment, then the control column can be left blank.
Otherwise, make sure that the values in control for experimental samples
correspond to a value in grouping which has been provided for the matched control.
For example, for the dataset described in original the CUT&Tag tutorial written by Ye Zheng, the annotated metadata should be set up as follows:
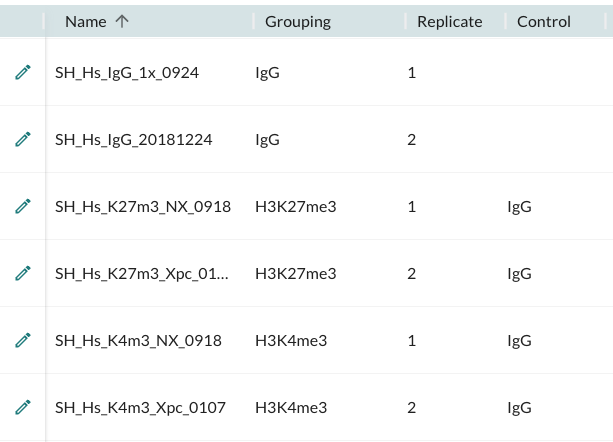
Note: If there are an equal number of replicates assigned to the samples from the control group as is the case above, the IgG controls will automatically be assigned to the same replicate number. If there is a mismatch then the first replicate of the control group will be assigned to all.
Parameters:
For an extended description of the parameters used for CUT&RUN, see the official parameter guide for nf-core/cutandrun (2.0).
Workflow Repository: github.com/nf-core/cutandrun
Citations:
- nf-core/cutandrun: Chris Cheshire, charlotte-west, nf-core bot, David Ladd, Chris Fields, Harshil Patel, Jordi Deu-Pons, Phil Ewels, & Kevin Menden. (2022). nf-core/cutandrun: nf-core/cutandrun v2.0 Copper Cobra (2.0). Zenodo. https://doi.org/10.5281/zenodo.6624266
- nf-core: Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020 Mar;38(3):276-278. doi: 10.1038/s41587-020-0439-x. PubMed PMID: 32055031.
- CUT&RUN: Meers, M. P., Bryson, T. D., Henikoff, J. G., & Henikoff, S. (2019). Improved CUT&RUN chromatin profiling tools. eLife, 8. https://doi.org/10.7554/eLife.46314
- CUT&Tag: Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., Ahmad, K., & Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications, 10(1), 1930. https://doi.org/10.1038/s41467-019-09982-5
- Additional references
ATAC-seq (nf-core/atacseq)
ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is a technique used in molecular biology to assess genome-wide chromatin accessibility. nf-core/atacseq is a best-practice bioinformatics analysis pipeline for the analysis of ATAC-seq data.

Source: Grandi, Modi, Kampman, and Corces. Nature Protocols. 2022
User Guide
Uploading Data:
Input data for the ATAC-seq pipeline should be provided as paired-end FASTQ files with filenames following the Illumina naming convention.
Parameters:
The most important parameters to understand for running ATAC-seq analysis are those which govern the way in which peaks are called. When reads are found within a peak, that indicates that chromatin is 'open' and transcriptionally active in that region. The algorithm used for identifying those peaks is MACS (Model-based Analysis for ChIP-Seq). For a discussion on the behavior and parameters for this algorithm please refer to this useful documentation.
For an extended description of the parameters available for customizing the ATAC-seq analysis, please refer to the official documentation for the nf-core/atacseq pipeline.
Workflow Repository: github.com/nf-core/atacseq
Citations:
- nf-core/atacseq: Harshil Patel, Phil Ewels, Jose Espinosa-Carrasco, Alexander Peltzer, Drew Behrens, Gisela Gabernet, Mingda Jin, Matthias Hörtenhuber, & Maxime U. Garcia. (2022). nf-core/atacseq: nf-core/atacseq v1.2.2 - Iron Ossifrage (1.2.2). Zenodo. https://doi.org/10.5281/zenodo.6544493
- ATAC-seq: Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013 Dec;10(12):1213-8. doi: 10.1038/nmeth.2688. Epub 2013 Oct 6. PMID: 24097267; PMCID: PMC3959825.
- ATAC-seq Protocol: Grandi FC, Modi H, Kampman L, Corces MR. Chromatin accessibility profiling by ATAC-seq. Nat Protoc. 2022 Jun;17(6):1518-1552. doi: 10.1038/s41596-022-00692-9. Epub 2022 Apr 27. PMID: 35478247; PMCID: PMC9189070.
- BWA: Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 Jul 15;25(14):1754-60. doi: 10.1093/bioinformatics/btp324. Epub 2009 May 18. PubMed PMID: 19451168; PubMed Central PMCID: PMC2705234.
- MACS2: Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. Epub 2008 Sep 17. PubMed PMID: 18798982; PubMed Central PMCID: PMC2592715.
- DEseq2: Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. PubMed PMID: 25516281; PubMed Central PMCID: PMC4302049.
ChIP-seq (nf-core/chipseq)
ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins (wikipedia). The nf-core/chipseq pipeline is a best practice workflow for the analysis of these datasets.
 Image credit: Jkwchui
(source)
Image credit: Jkwchui
(source)
{kind=link}
User Guide
Sample Metadata: Control Annotation (e.g. IgG):
If you feel comfortable with JSON files, you can follow the instructions below to edit the JSON schemas for metadata. If not, contact the Cirro team for assistance.
After uploading paired-end DNA sequencing data in FASTQ format, samples must be annotated by (1) the antibody used for ChIP, (2) the matched control sample, and (3) the replicate. Each non-control group must be matched with the appropriate control group for proper normalization and peak detection.
To indicate the appropriate sample groups, first add fields for group, antibody, control, and
replicate to the metadata.schema.json for the project as shown here:
{
"$schema": "http://json-schema.org/draft-07/schema#",
"type": "object",
"properties": {
"group": {
"type": "string",
"description": "Used to indicate groups of samples which are replicates of the same experimental condition"
},
"antibody": {
"type": "string",
"description": "(optional) Used to indicate the antibody used for ChIP"
},
"replicate": {
"type": "integer",
"description": "Used to indicate the experimental replicate within each experimental condition"
},
"control": {
"type": "string",
"description": "Used to indicate which 'group' should be used as a matched control"
}
}
}
Next, update those metadata fields for the uploaded data using the sample annotation tool provided in Cirro.
Make sure that the value in the
controlcolumn matches the value in thegroupcolumn for the appropriate control.
A longer description of the sample metadata can be found on the documentation page
for nf-core/chipseq.
Note that fastq_1 and fastq_2 need not be provided, as they will be filled in
appropriately for each sample by Cirro directly.
Parameters:
For an extended description of the parameters used for ChIP-seq, see the official parameter guide for nf-core/chipseq (1.2.2).
Workflow Repository: github.com/nf-core/chipseq
Citations:
- nf-core/chipseq: Harshil Patel, Chuan Wang, Phil Ewels, Tiago Chedraoui Silva, Alexander Peltzer, Drew Behrens, Maxime Garcia, mashehu, Rotholandus, Sofia Haglund, & Winni Kretzschmar. (2021). nf-core/chipseq: nf-core/chipseq v1.2.2 - Rusty Mole (1.2.2). Zenodo. https://doi.org/10.5281/zenodo.4711243
- nf-core: Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020 Mar;38(3):276-278. doi: 10.1038/s41587-020-0439-x. PubMed PMID: 32055031.
- BWA: Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 Jul 15;25(14):1754-60. doi: 10.1093/bioinformatics/btp324. Epub 2009 May 18. PubMed PMID: 19451168; PubMed Central PMCID: PMC2705234.
- MACS2: Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. Epub 2008 Sep 17. PubMed PMID: 18798982; PubMed Central PMCID: PMC2592715.
- HOMER: Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010 May 28;38(4):576-89. doi: 10.1016/j.molcel.2010.05.004. PubMed PMID: 20513432; PubMed Central PMCID: PMC2898526.
- DEseq2: Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. PubMed PMID: 25516281; PubMed Central PMCID: PMC4302049.
Methylation (Bisulfite-Sequencing) - nf-core/methylseq
Bisulfite sequencing is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity (WikiPedia).
The nf-core/methylseq workflow analyzes raw FASTQ files with either the Bismark or bwa-meth / methyldactyl analysis tools.
User Guide
To analyze a dataset with the nf-core/methylseq pipeline, simply upload paired-end FASTQ files and select the "methylseq (nf-core)" analysis tool.
Depending on the method which was used for library preparation, you may select from the preset analysis settings for:
- PBAT
- MspI
- SLAM-seq
- EM-seq
- Single-cell bisulfite sequencing
- Accel kit
- CEGX bisulfite kit
- Epignome kit
- Zymo kit
A full description of the parameters available for the analysis can be found in the official nf-core/methylseq documentation.
Workflow Repository: github.com/nf-core/methylseq
Citations:
- nf-core/methylseq: Phil Ewels, Rickard Hammarén, Alexander Peltzer, phue, Sven F., Paolo Di Tommaso, Maxime Garcia, Johannes Alneberg, Andreas Wilm, & Alessia. (2019). nf-core/methylseq: nf-core/methylseq version 1.3 (1.3). Zenodo. https://doi.org/10.5281/zenodo.2555454
- Bismark: Felix Krueger, Simon R. Andrews, Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications, Bioinformatics, Volume 27, Issue 11, 1 June 2011, Pages 1571–1572, https://doi.org/10.1093/bioinformatics/btr167
- bwa-meth: Pedersen, Brent S., et al. "Fast and accurate alignment of long bisulfite-seq reads." arXiv preprint arXiv:1401.1129 (2014).
- MethylDackel: https://github.com/dpryan79/methyldackel
Scrub Human Sequences
When submitting genomic sequences to public repositories, it is important to remove any human sequences which may have been inadvertently included. This is particularly needed for specimens which are obtained from a human source, but for which the primary organisms of interest are non-human (for example, when studying the human microbiome).
This workflow will use the NCBI-approved tool for masking all human sequences with N's in the raw FASTQ data. While this can be used to scrub previously-analyzed datasets in preparation for submission to public repositories (as is required for the Sequence Read Archive), it could also be used to scrub datasets at the start of a project prior to running any analyses.
Supports combining input datasets in a single analysis.
User Guide
Workflow Repository: github.com/FredHutch/sra-human-scrubber-nf
Tool Repository: github.com/ncbi/sra-human-scrubber
Citations:
- SRA Taxonomy Analysis Tool: Katz, K.S., Shutov, O., Lapoint, R. et al. STAT: a fast, scalable, MinHash-based k-mer tool to assess Sequence Read Archive next-generation sequence submissions. Genome Biol 22, 270 (2021). https://doi.org/10.1186/s13059-021-02490-0
GATK: Convert paired FASTQ to uBAM
The Genome Analysis Toolkit (GATK) is a broadly-used set of utilities for analyzing genome sequence data, including the identification of germline and somatic variations.
Input genome sequence data for GATK utilities are expected to be formatted as unmapped BAM files. A discussion of the utility of the BAM file format can be found on the GATK user forum. This utility provides the ability to convert paired-end FASTQ data into unmapped BAM files which can then be processed further by GATK tools.
Workflow Repository: github.com/gatk-workflows/seq-format-conversion
GATK: Variant Discovery Pre-Processing
The processing-for-variant-discovery-gatk4 WDL pipeline implements data pre-processing according to the GATK Best Practices. The workflow takes as input an unmapped BAM list file (text file containing paths to unmapped bam files) to perform preprocessing tasks such as mapping, marking duplicates, and base recalibration. It produces a single BAM file and its index suitable for variant discovery analysis using tools such as Haplotypecaller.
Workflow Repository: github.com/gatk-workflows/gatk4-data-processing
GATK: Germline Short Variant Discovery
The Germline Variant Discovery workflow runs the GATK4 HaplotypeCaller tool in GVCF mode on a single sample according to GATK Best Practices. When executed the workflow scatters the HaplotypeCaller tool over the input bam sample using an interval list file. The output produced by the workflow will be a single GVCF file which can then be provided to GenomicsDBImport along with several other GVCF files to call for variants simultaneously, producing a multisample VCF.
Workflow Repository: github.com/gatk-workflows/gatk4-germline-snps-indels
GATK: Whole Genome Germline Single Sample
The Whole Genome Germline Single Sample (WGS) pipeline implements data pre-processing and initial variant calling according to the GATK Best Practices for germline SNP and Indel discovery in human whole-genome sequencing data. It includes the DRAGEN-GATK mode, which makes the pipeline functionally equivalent to DRAGEN’s analysis pipeline (read more in this DRAGEN-GATK blog).
The GATK Whole Genome Germline Single Sample workflow supports DRAGEN analysis in two modes:
- Functional Equivalence: outputs are functionally equivalent to those produced with the DRAGEN hardware, and
- Maximum Quality: uses the DRAGMAP aligner and DRAGEN variant calling, but with additional parameters that produce maximum quality results that are not functionally equivalent to the DRAGEN hardware.
More details on the implementation of DRAGEN are provided by the workflow documentation.
Supports combining input datasets in a single analysis.
Workflow Repository: https://github.com/broadinstitute/warp
Workflow Release: WholeGenomeGermlineSingleSample_v3.1.6