Pipeline Catalog: Proteomics
Uploading Data
Spectral data generated from proteomics experiments can be uploaded in either
mzML or
RAW
file format.
In addition, the experimental design structure which describes that set of files should be
uploaded as a samplesheet.csv for each dataset.
Note: The path for each of the spectra files in the
samplesheet.csvshould not include any parent folders. For example, instead of/path/to/dataset/Sample1.mzML, just useSample1.mzML.
Reference Data - Genome
The set of protein targets used for analyzing peptide spectra must be provided as a
Reference Genome (FASTA) on the References page.
Note: If the protein FASTA provided as a reference does not already contain 'decoy' sequences, then the "Add Decoys" option must be selected when running the analysis.
Data-Dependent Acquisition – Label Free Quantitation (DDA-LFQ)
Analysis of DDA-LFQ data is performed using the nf-core/quantms pipeline.
Input Data:
Input files can be processed from either the .mzML or .raw file formats.
The input for this workflow should be uploaded as either the "Proteomics (RAW)"
or "Proteomics (mzML)" dataset type.
Include a full batch of samples to be processed as a single dataset for optimal
processing.
Supports combining input datasets in a single analysis.
Sample Metadata:
In addition to the spectral data files, metadata may be provided to annotate
specific details of the experimental design.
To provide sample-level metadata appropriately,
upload a samplesheet.csv
as part of the dataset.
The metadata included in the samplesheet.csv mirrors the
Sample and Data Relationship (SDRF) Format for Proteomics,
with the important distinction that the samplesheet.csv must be comma-delimited
(and not tab-delimited as expected for SDRF).
Required Metadata Fields:
When providing a samplesheet.csv the columns sample and file must be provided.
Every sample in the dataset must be listed on its own line.
If there are multiple replicate files from the same sample, use additional columns
file_2, file_3, etc.
Note: The
filecolumn should list the relative path of the file within the uploaded folder, not the full URI.
The information in those two columns is used to automatically populate the columns:
- source name (from
sample) - comment[file uri] (from
file) - comment[data file] (from
file)
If no samplesheet.csv is provided the following fields will be populated
with default values:
- characteristics[organism]: Homo sapiens
- characteristics[organism part]: not applicable
- characteristics[disease]: not applicable
- characteristics[cell type]: not applicable
- characteristics[biological replicate]: not applicable
- comment[fraction identifier]: 1
- comment[technical replicate]: (autoincrement files with the same sample)
- comment[cleavage agent details]: NT=Trypsin
- comment[instrument]: NT=LTQ Orbitrap XL
- comment[label]: NT=label free sample
Optional Metadata Fields:
The following fields may also be provided as columns in the samplesheet.csv,
with the values being passed along to the workflow for processing:
- characteristics[cell line]
- characteristics[ancestry category]
- characteristics[age]
- characteristics[sex]
- characteristics[developmental stage]
- characteristics[individual]
- material type
- technology type
- comment[flow rate chromatogram]
- comment[gradient time]
- comment[fractionation method]
- comment[modification parameters]
- comment[modification parameters]
- comment[modification parameters]
- comment[fragment mass tolerance]
- comment[precursor mass tolerance]
- factor value[flow rate chromatogram]
- factor value[gradient time]
Analyze Subset of Files:
If you wish to analyze only a portion of the files in a dataset, use the "Analyze Subset" dropdown menu when launching the workflow. If no files are selected (the default), then everything present in the dataset will be analyzed. If any files are selected, then only those files will be analyzed.
Workflow Overview:
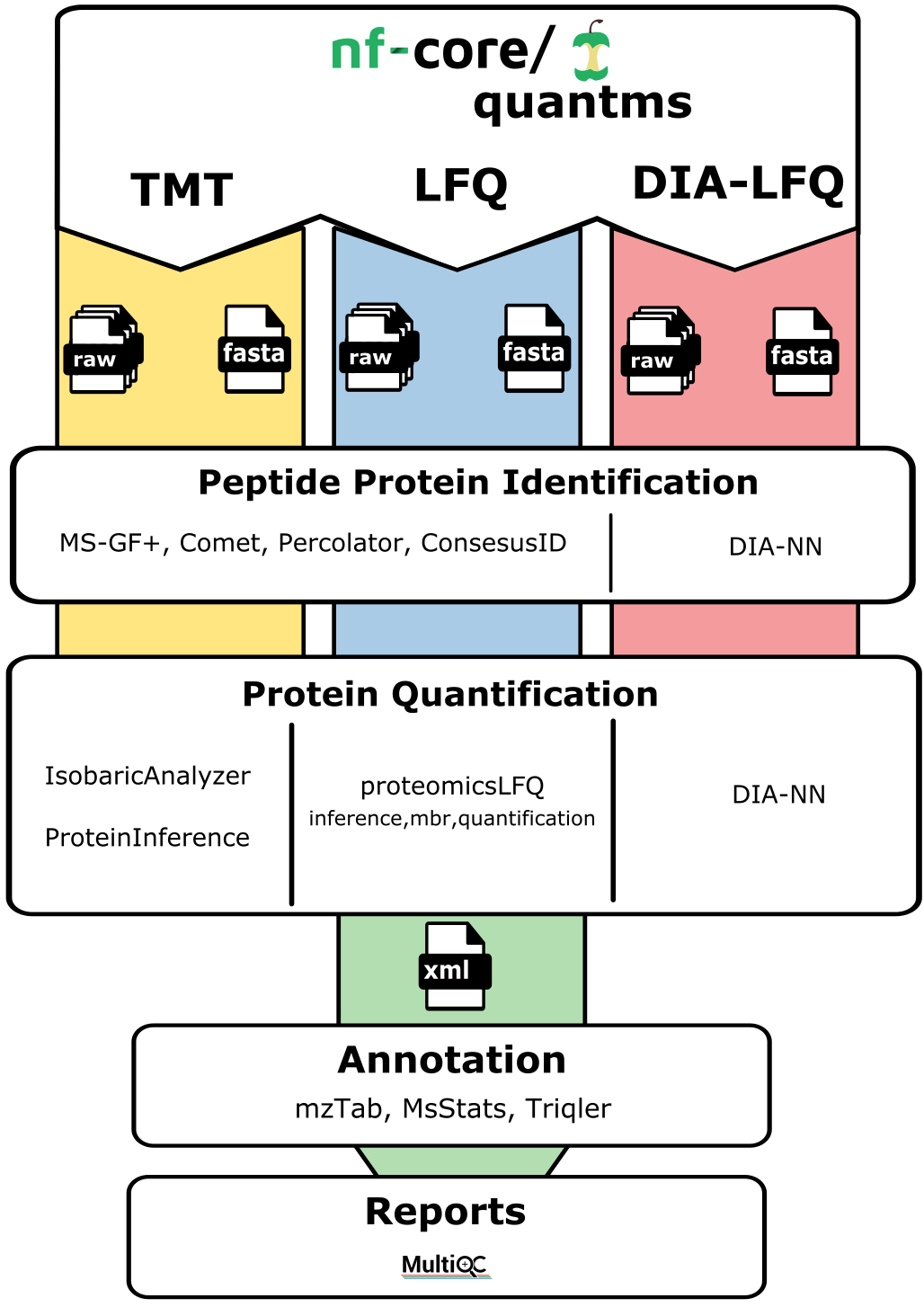
While the nf-core/quantms workflow supports a variety of data types (DDA-LFQ, DDA-Isobaric, and DIA-LFQ), each of those approaches is provided as a distinct pipeline within Cirro.
For more details on the parameters available, see the full workflow documentation.
Note: The DDA-LFQ pipeline in Cirro is run with the parameters:
acquisition_method: "dda", labelling_type: "label free sample"
Quality Control Report (nf-core/quantms)
To generate a high-level summary of the results generated by the nf-core/quantms pipeline, this workflow summarizes the number of spectra, peptides, and proteins which have been detected.
Quality control metrics are generated at the dataset level, combining results across all of the individual files which have been analyzed in a batch.
To compare across runs, the quality control report can be executed on multiple datasets. The results from each dataset will be labeled with the name assigned by the user to the dataset. Even if that dataset name has been modified after creation, the name at the of QC report creation will be used to label the results.
Results include:
- Num_Proteins_Identified
- Num_Proteins_Quantified
- Num_MS2_Spectra_Identified
- Num_Peptides_Identified
In addition, plots are provided in HTML, PDF, and PNG format showing the distribution of results across the datasets.