Preprocess Script
The preprocess script is an optional script used to provide inputs to a workflow which cannot be solely formatted by the analysis submission form. This can help with common tasks like:
- Preparing the essential
inputs.jsonandoptions.jsonfor WDL/Cromwell workflows - Formatting a
samplesheet.csvfor Nextflow workflows - Performing any validation of files or parameters before the workflow is run
- Executing arbitrary logic based on user inputs
The flow of an analysis in Cirro with a preprocess script is as follows:
- User uploads a dataset with input files and sample metadata (
samplesheet.csv). - Cirro reads the
samplesheet.csvfile and stores the metadata in the database. - User selects a pipeline, enters parameters, and submits the analysis.
- Cirro writes the parameters and sample metadata and makes them available to the preprocess script as ds.params, ds.samplesheet, and ds.files.
- Cirro runs the user-provided preprocess script, and writes out any additional files (if specified).
- Cirro runs the workflow.
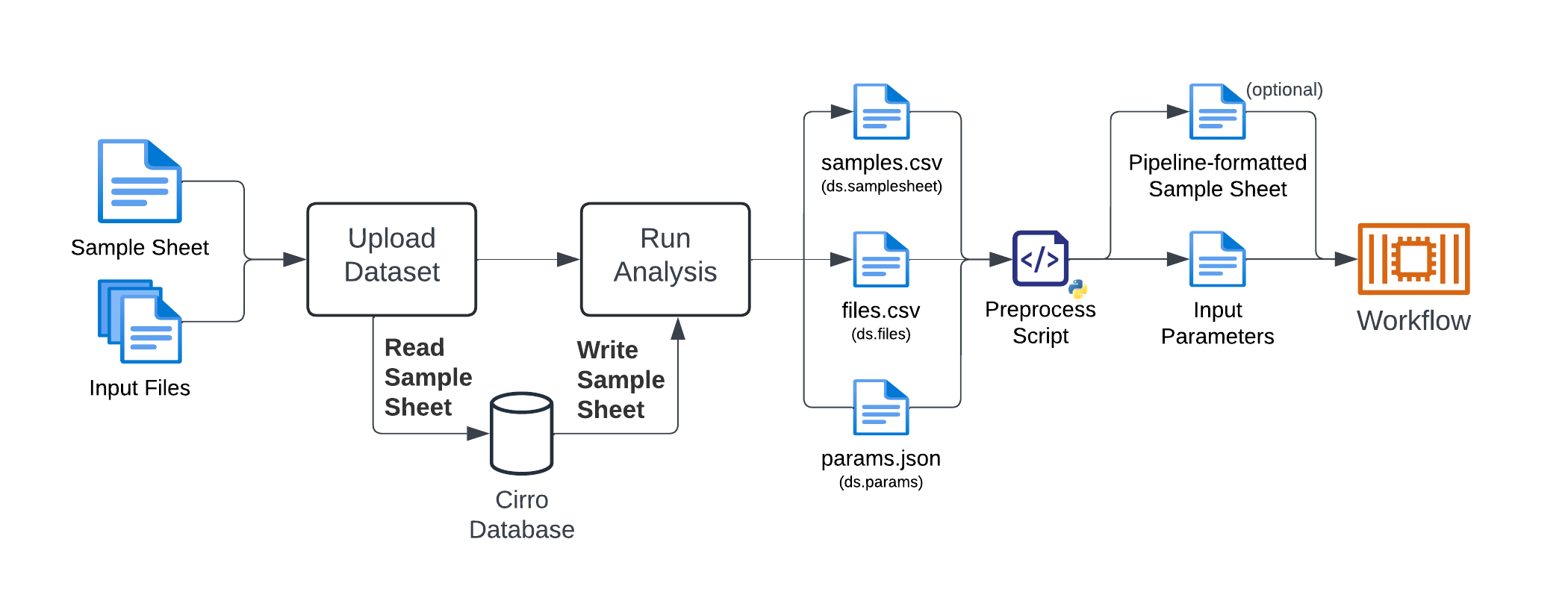
For workflows that require samplesheet-style inputs, the preprocess script is necessary because:
- At the time of upload, the file locations are not known. The
ds.filestable will contain the S3 paths to the files needed by the workflow. - The sample metadata information may have been changed by the Samples table after upload.
- The format of the samplesheet that the user uploads is decoupled the samplesheet that the workflow expects.
- The user uploading the dataset might not be aware of the specific format required by the workflow.
- The user might want to run multiple workflows on the same dataset, each with a different samplesheet format.
- The user might want to run the same workflow on multiple input datasets, combining them into a single samplesheet.
When developing this script, it is recommended to use the Developer Tools to generate the PreprocessDataset class for testing (as you will not be able to call the from_running method during local development).
Accessing Dataset Information
Each preprocess script will typically start with:
from cirro.helpers.preprocess_dataset import PreprocessDataset
ds = PreprocessDataset.from_running()
The ds object contains all of the information needed for the dataset
which has been created for the analysis results.
Explanation: When a Pipeline is run by a user, a new Dataset is created to hold the results of that analysis. The new dataset is populated with
- the location of the input data
- the parameters selected by the user
- the sample metadata (a snapshot from the time of submission)
The attributes of the ds object which can be used in the preprocess
script are:
ds.files
A pandas DataFrame containing information on the files contained in the input datasets, and the sample that each file is assigned to.
Any other metadata which is assigned at the file level (e.g.,
the read variable which is parsed for paired-end FASTQ data)
will also be present in ds.files.
print(ds.files)
| sample | file | sampleIndex | read |
|---|---|---|---|
| sampleA | s3://path/to/sampleA_R1.fastq.gz | 1 | 1 |
| sampleA | s3://path/to/sampleA_R2.fastq.gz | 1 | 2 |
| sampleB | s3://path/to/sampleB_R1.fastq.gz | 2 | 1 |
| sampleB | s3://path/to/sampleB_R2.fastq.gz | 2 | 2 |
Note: The
sampleIndexis used to differentiate between files which are uploaded as a pair, but which have the same sample name (e.g., combining FASTQ files from multiple lanes of a genome sequencer).
ds.samplesheet
A pandas DataFrame containing all of the metadata assigned to the samples present in the input datasets (at the time of analysis).
print(ds.samplesheet)
| sample | group | age |
|---|---|---|
| sampleA | Group_1 | 55 |
| sampleB | Group_2 | 72 |
ds.pivot_samplesheet()
Pivot the long-form ds.files to a wide-form DataFrame and combines the sample metadata from ds.samplesheet.
The arguments required are:
index(default["sampleIndex", "sample", "lane"]): The columns used for the index in the wide samplesheet. If any of these columns do not exist inds.files.columns, they will be silently omitted from the results.pivot_columns(defaultread): The column containing values which will be used for column headers in the wide samplesheetmetadata_columns(defaultNone): The columns which will be included in the wide samplesheet. By default, all columns inds.samplesheetwill be included.column_prefix(defaultfastq_): The string prefix will be added to the values from thepivot_columnsfile_filter_predicate(defaultNone): A function which takes a row of theds.filesDataFrame and returnsTrueif the file should be included in the wide samplesheet, orFalseif it should be excluded.
For example, using the ds.files table above:
ds.pivot_samplesheet()
| sampleIndex | sample | fastq_1 | fastq_2 | group | age |
|---|---|---|---|---|---|
| 1 | sampleA | s3://path/to/sampleA_R1.fastq.gz | s3://path/to/sampleA_R2.fastq.gz | Group_1 | 55 |
| 2 | sampleB | s3://path/to/sampleB_R1.fastq.gz | s3://path/to/sampleB_R2.fastq.gz | Group_2 | 72 |
Note that the results of
ds.pivot_samplesheet()may often reproduce the contents of thesamplesheet.csvused to upload a paired-end FASTQ dataset. Importantly, the path to the individual files will reflect their current location in Cirro, and not the relative path they were uploaded from.
ds.metadata
Detailed information on the dataset which has been created for the analysis results.
{
"dataset": {
"id": "111",
"createdAt": "2024-09-23T19:55:28.020052Z",
"updatedAt": "<SNIP>",
"createdBy": "hello@cirro.bio",
"params": {
"level": 3,
"experiment_format": {
"plates": "A B C",
"wells": "1 2 3"
},
"custom_options": {
"with_custom": false
}
},
"info": {},
"projectId": "000",
"tags": [],
"processId": "process-hutch-interop-qc-1_0",
"name": "New Dataset",
"status": "PENDING",
"s3": "s3://project-000/datasets/111",
"dataPath": "s3://project-000/datasets/111/data"
},
"project": {
"id": "000",
"name": "Test Project",
"description": "Test Project Description",
"status": "COMPLETED"
},
"process": {
"id": "process-hutch-interop-qc-1_0",
"createdAt": "<SNIP>",
"updatedAt": "<SNIP>",
"pipelineType": "Community",
"name": "InterOp QC",
"description": "InterOp QC Reporting - Generate QC metrics from Illumina Sequencing run",
"executor": "NEXTFLOW",
"documentationUrl": "https://github.com/FredHutch/interop-nf",
"code": {
"repositoryType": "GITHUB_PUBLIC",
"uri": "FredHutch/interop-nf",
"version": "1.0",
"script": "main.nf"
},
"archived": false,
"category": "Quality Control"
},
"inputs": [
{
"id": "aaa",
"createdAt": "<SNIP>",
"updatedAt": "<SNIP>",
"createdBy": "hello@cirro.bio",
"params": {},
"info": {},
"projectId": "000",
"sourceDatasetIds": [],
"tags": [],
"processId": "sequencing-run",
"name": "MiSeq Demo 1",
"status": "COMPLETED",
"description": "test",
"s3": "s3://project-000/aaa",
"dataPath": "s3://project-000/aaa/data"
},
{
"id": "bbb",
"createdAt": "<SNIP>",
"updatedAt": "<SNIP>",
"createdBy": "hello@cirro.bio",
"params": {},
"info": {},
"projectId": "000",
"sourceDatasetIds": [],
"tags": [],
"processId": "sequencing-run",
"name": "MiSeq Demo 2",
"status": "COMPLETED",
"description": "test",
"s3": "s3://project-000/bbb",
"dataPath": "s3://project-000/bbb/data"
}
]
}
ds.params
A dictionary
with all of the parameter values populated by user input using the process-form.json
and process-input.json configurations.
This object is only used to access parameter values (e.g. ds.params['foo'] or
ds.params.get('foo')).
Adding or removing parameters from this object will not be carried through to
the workflow.
For that purpose, use ds.add_param or ds.remove_param.
ds.references_base
S3 base path to the Cirro shared references bucket.
ds.add_param()
Method to add a parameter using its keyword and value.
If the parameter keyword is already in use, this method
will raise an error unless overwrite=True is set.
# To add the parameter "new_param", with value 1
ds.add_param("new_param", 1)
ds.params["new_param"] == 1
Note: While
ds.params["new_param"] = 1would assign the value within the context of the preprocess script, that value would not be passed along to the workflow without using the.add_parammethod.
ds.remove_param()
Method to remove a parameter from the workflow parameters.
If the parameter does not currently exist, this method
will raise an error unless force=True is set.
# To remove the parameter "new_param"
ds.remove_param("new_param")
ds.params.get("new_param") is None
ds.logger.info()
Print a helpful message to the logs, which is very helpful for debugging purposes.
# For example, to get an idea of what the samplesheet
# looks like before running the workflow
samplesheet = ds.wide_samplesheet()
ds.logger.info(samplesheet.to_csv(index=None))
WDL / Cromwell
For a WDL pipeline to run in Cirro (which uses the Cromwell executor in 'run mode'),
at least two files must be created by the preprocess script:
inputs.*.json and options.json.
Differentiating 'inputs' and 'options'
While Cirro sets up a single set of 'parameters' for a workflow,
the configuration of the Cromwell executor will expect some of
those values to be provided as options while others are inputs.
For example, the location where final output files will be written
is typically provided as final_workflow_outputs_dir (which is an option),
while the reference genome used for alignment may be provided as
WholeGenomeGermlineSingleSample.references (which is an input).
For this reason, the preprocess script is used to differentiate the
parameters which should be submitted as inputs versus options.
After the preprocess script has completed, the headnode will expect to find:
- A file named
options.json - One or more files named
inputs.*.json
Execution of Multiple inputs.json
The philosophy of WDL workflows established by GATK is that a workflow processes a single sample (or panel of samples) at a time. In contrast, the approach of nf-core is often that a batch of samples can be processed in parallel.
To support both approaches for WDL (analyzing either a single or multiple
samples), the Cirro headnode can run the specified workflow multiple times,
once for each file encountered with the file name pattern inputs.*.json
(e.g. inputs.1.json, inputs.2.json) (note that * can be any string,
not just numerals).
Because the Cromwell executor is used in "run mode", each file of inputs will be processed in serial (within the analysis of a single Cirro Dataset). While this approach may be slower in some cases than parallel execution, one minor benefit is that any computational steps which are repeated across runs will be cached, and will not be unnecessarily repeated during the workflow execution.
Useful Patterns
Splitting up inputs and options using a keyword prefix
A convenient approach for differentiating between workflow parameters
which should be supplied via inputs vs. options is the presence
of a keyword prefix which is named for the workflow being executed.
For example, when running the workflow named WholeGenomeGermlineSingleSample,
all of the input parameters will be named something starting with
WholeGenomeGermlineSingleSample.*.
To split up parameters based on the presence of that prefix and write out the appropriate files:
from cirro.helpers.preprocess_dataset import PreprocessDataset
import json
# Get the workflow execution information
ds = PreprocessDataset.from_running()
# Name of the WDL workflow being run
WORKFLOW_PREFIX = "WholeGenomeGermlineSingleSample"
# Split up the inputs and options using dict comprehension
inputs = {
kw: val
for kw, val in ds.params.items()
if kw.startswith(WORKFLOW_PREFIX)
}
options = {
kw: val
for kw, val in ds.params.items()
if not kw.startswith(WORKFLOW_PREFIX)
}
# Write out the inputs and options
json.dump(inputs, open("inputs.1.json", "w))
json.dump(options, open("options.json", "w))
Apply default params:
# Boilerplate which can be used with all workflows
ds.add_param("use_relative_output_paths", True)
ds.add_param("read_from_cache", True)
ds.add_param("write_to_cache", True)
Nextflow
The preprocess script is technically optional for Nextflow workflows, but it is often required for nf-core style workflows in which inputs are formatted as a samplesheet.
By default, all of the parameters set up in the pipeline configuration
will be passed to a Nextflow workflow as parameters (in the params scope).
As such, any modifications made to the parameters using ds.add_param or
ds.remove_param will be reflected in the inputs to Nextflow without having
to write any other files to the working directory.
Useful Patterns
Remove any index reads (for paired-end FASTQ datasets with I1/I2 in addition to R1/R2):
# Filter out any index files that may have been uploaded
ds.files = ds.files.loc[
ds.files.apply(
lambda r: r.get('readType', 'R') == 'R',
axis=1
)
]
Set up a wide-form samplesheet for paired-end FASTQ inputs, including metadata:
# Make a wide-form samplesheet with columns: sample, fastq_1, and fastq_2
samplesheet = ds.pivot_samplesheet()
# Save to a file
samplesheet.to_csv("samplesheet.csv", index=None)
# Set up a workflow param pointing to that file (e.g., for nf-core/rnaseq)
ds.add_param("input", "samplesheet.csv")
Unit Testing
When developing the preprocess.py script, it can be helpful to test the script locally using a unit testing framework, rather than running the entire Cirro analysis workflow (which can be time-consuming).
To do this, you will need to make sure that your preprocess.py script is written in a way that allows you to test it in isolation.
For example, if you need to create a sample sheet, you should extract the logic for creating the sample sheet into a separate function that can take the samples and files as arguments.
Here is an example of how you might structure your preprocess.py script to allow for unit testing:
import pandas as pd
from cirro.helpers.preprocess_dataset import PreprocessDataset
def make_manifest(samples: pd.DataFrame, files: pd.DataFrame) -> pd.DataFrame:
"""
Create a samplesheet for my workflow
"""
# Your logic for creating the samplesheet goes here
pass
if __name__ == '__main__':
ds = PreprocessDataset.from_running()
manifest = make_manifest(ds.samplesheet, ds.files)
manifest.to_csv('samplesheet.csv', index=False)
You can then create a separate test script, preprocess_test.py, that imports the make_manifest function and tests it using a unit testing framework like unittest.
Here is an example of how you might structure your preprocess_test.py script:
import unittest
from io import StringIO
import pandas
from preprocess import make_manifest
class PreprocessTest(unittest.TestCase):
def test_formats_samplesheet(self):
manifest = make_manifest(
samples=pandas.read_csv("<TEST_FILE_PATH>"),
files=pandas.read_csv("<TEST_FILE_PATH>")
)
manifest_csv = manifest.to_csv(index=False).strip()
expected_manifest_csv = pandas.read_csv("<EXPECTED_MANIFEST_FILE_PATH>").to_csv(index=False).strip()
self.assertEqual(manifest_csv, expected_manifest_csv)
In this test script, you would replace the file path placeholders to your test files.
You can also use the StringIO class to create in-memory CSV data for testing.
Use the generate_samplesheets_for_dataset method to generate this test data based on actual Cirro data.
More info on the Integrating Pipelines notebook.
Full Example
Let's say we have a Nextflow workflow that requires a sample sheet to be in this format:
| sample | forwardReads | reverseReads | group |
|---|---|---|---|
| sampleA | s3://path/to/sampleA_R1.fastq.gz | s3://path/to/sampleA_R2.fastq.gz | Group_1 |
| sampleB | s3://path/to/sampleB_R1.fastq.gz | s3://path/to/sampleB_R2.fastq.gz | Group_2 |
The script will need make the following changes to the Cirro-provided sample sheet:
- filter out any index files that may be present in the dataset upload,
- pivot the files table to have one row per sample, with columns for each read,
- merge the samples and files tables to bring in additional metadata,
- rename the sample column to sampleID and read number columns to forwardReads and reverseReads,
- drop all unnecessary columns,
- make sure the sampleID only has allowed characters,
- check to make sure there are samples in the dataset.
Expand the section below to see the full preprocess.py script:
preprocess.py
import re
import pandas
import pandas as pd
from cirro.helpers.preprocess_dataset import PreprocessDataset
def make_manifest(samples: pandas.DataFrame, files: pandas.DataFrame) -> pandas.DataFrame:
"""
Create a samplesheet for my workflow
"""
# Filter out any index files that may be present, we only want the reads
files = files[files['readType'] == 'R']
# Pivot the files table to have one row per sample, with columns for each read
files = pd.concat([
pd.DataFrame(dict(
sample=sample,
**{
f"R{read}": read_files['file'].sort_values().tolist()
for read, read_files in sample_files.groupby("read")
}
))
for sample, sample_files in files.assign(
read=files["read"].apply(int)
).query(
"read <= 2"
).groupby("sample")
])
# Merge samples and files tables
samplesheet = pd.merge(files,
samples,
left_on="sample",
right_on="sample",
validate="1:1",
how="inner")
# Rename sample column to sampleID and read number columns
samplesheet = samplesheet.rename(columns={
"sample": "sampleID",
"R1": "forwardReads",
"R2": "reverseReads"
})
# Drop all columns except sampleID, forwardReads, reverseReads, and group
samplesheet = samplesheet[["sampleID", "forwardReads", "reverseReads", "group"]]
# Make sure the sampleID only has allowed characters (remove non-alphanumeric characters)
samplesheet["sampleID"] = samplesheet["sampleID"].apply(
lambda s: re.sub('\W+', '_', str(s))
)
if samplesheet.shape[0] == 0:
raise ValueError("No samples found in the dataset")
return samplesheet
if __name__ == '__main__':
ds = PreprocessDataset.from_running()
manifest = make_manifest(ds.samplesheet, ds.files)
manifest.to_csv('samplesheet.csv', index=False)
To test this script, we create the file preprocess_test.py in the same directory as preprocess.py.
See the full test script below:
preprocess_test.py
import unittest
from io import StringIO
import pandas
from preprocess import make_manifest
TEST_SAMPLESHEET = """
sample,group,randomData
sample1,control,test
sample2,treatment,test
""".strip()
TEST_FILES = """
sample,file,readType,read
sample1,sample1_R1.fastq.gz,R,1
sample1,sample1_R2.fastq.gz,R,2
sample1,sample1_I1.fastq.gz,I,1
sample2,sample2_R1.fastq.gz,R,1
""".strip()
EXPECTED_MANIFEST = """
sampleID,forwardReads,reverseReads,group
sample1,sample1_R1.fastq.gz,sample1_R2.fastq.gz,control
sample2,sample2_R1.fastq.gz,,treatment
""".strip()
class PreprocessTest(unittest.TestCase):
def test_formats_samplesheet(self):
manifest = make_manifest(
samples=pandas.read_csv(StringIO(TEST_SAMPLESHEET)),
files=pandas.read_csv(StringIO(TEST_FILES))
)
manifest_csv = manifest.to_csv(index=False).strip()
self.assertEqual(manifest_csv, EXPECTED_MANIFEST)
def test_throws_error_no_samples(self):
files = pandas.read_csv(StringIO(TEST_FILES))
only_index_files = files[files['readType'] == 'I']
with self.assertRaises(ValueError):
make_manifest(
samples=pandas.read_csv(StringIO(TEST_SAMPLESHEET)),
files=only_index_files
)